
Guidance of CCMS
Greetings

Supercomputing System for Computational Materials Science
Mission of Center for Computational Materials Science
System Overview
System Configuration
MASAMUNE-II consists of the Supercomputer (HPE Cray XD220v, HPE Scale-up Server 3200, and HPE Cray XD670). It is integrated by Hitachi, Ltd. The system specifications are shown below.

| System Name | Supercomputer | ||
|---|---|---|---|
| Model | HPE Cray XD220v | HPE Scale-up Server 3200 | HPE Cray XD670 |
| total nodes | General node : 120 Large memory node : 7 |
2 nodes | 11 nodes |
| CPU |
Intel Xeon Platinum 8480+
|
Intel Xeon Platinum 8490H
|
Intel Xeon Platinum 8480+
|
| Accelerator | - | - |
NVIDIA H100 80GB SXM5
|
| memory size | 512 GiB/node 2.0 TiB/node |
4.0 TiB/node | 1.0 TiB/node |
| Theoretical peak performance | 4.052 PFLOPS (CPU : 1.104 PFLOPS, GPU : 2.948 PFLOPS) |
||
Nickname and Front Panel Design of Supercomputing System
Nickname: MASAMUNE-II
MAterials science Supercomputing system for Advanced MUlti-scale simulations towards NExt-generation - II

Nickname “MASAMUNE-II” has been given to the supercomputing system of CCMS, continuing the nickname of the previous system “MASAMUNE-IMR”, in honor of “Masamune Date”, the first lord of Sendai in the 17th century, who dispatched the sailing ship “San Juan Bautista” towards the world from Sendai in 1613. The nickname “MASAMUNE-II” expresses our hope that creative and innovative achievements of the advanced multi-scale simulations on materials science by our supercomputing system will give great impacts towards the next-generation from Sendai to the World. “Masamune Date” is drawn on the front panel of our supercomputer by Sumi-E Artist OKAZU with the brave letter “弐” meaning “second” in Japanese Kanji. This “Masamune Date” on the front panel is looking ahead to the future advances in the materials science of the world from Sendai.
Achievement
Elucidation of Effect of Environments on Wear Phenomena of Diamond-like Carbon and Proposal of Design Princples towards Improving its Durability
Diamond-like carbon is an amorphous carbon material which has excellent properties such as ultra-low friction, wear resistance, and chemical stability and then it is expected to be used in space stations, airplane engines, drones, medical equipment, etc. However, the wear of diamond-like carbon can lead to reduced material lifespan, machine failure, and even unexpected accidents. Therefore, minimizing its wear to the utmost is a critical challenge for realizing a safe and secure society. Especially, its wear behavior varies signi cantly even with slight environmental differences such as partial pressures of water vapor, oxygen, nitrogen, and hydrogen, and then experimental research has not yet advanced our understanding of its underlying mechanism. Therefore, it is strongly demanded to utilize computational science simulations for clarifying the effects of environments and atmosphere on its wear mechanism and then for establishing the design guidelines to reduce its wear. However, unlike “material-intrinsic properties” such as hardness and electrical conductivity, the changes in wear phenomena due to the environments represent “material response characteristics”.
Therefore, understanding multi-physics phenomena including chemical reactions, friction, impact, stress, fluids, and heat transfer by computational science simulations is essential, which is one of the challenging tasks in computational science simulations. Therefore, by developing a reactive molecular dynamics simulator capable of elucidating complex multi-physics phenomena involving chemical reactions on the supercomputer “MASAMUNE-IMR,” for the first time in the world we discovered that wear occurs via two mechanisms, chemical wear and mechanical wear, on diamond-like carbon and revealed that the chemical reactions occurring on the surface of diamond-like carbon during friction vary depending on the environments such as water and oxygen, leading to differences in the form and amount of chemical wear and mechanical wear. This achievement contributes to extending the lifespan of mechanical systems as well as preventing failures and accidents.

Fig. Friction simulation of diamond-like carbon under water environment (Water hidden for clarity). We have demonstrated that water molecules dissociate into H+ and OH- at carbon atoms with dangling bonds on diamond-like carbon surfaces, initiating chemical wear where oxygen-containing organic molecules such as methanol and ethanol evaporate.
Jing Zhang, Yang Wang, Qian Chen, Yixin Su, Shandan Bai, Yusuke Ootani, Nobuki Ozawa, Koshi Adachi and Momoji Kubo
Carbon, 231 (2025) Art.No.119713, DOI: https://doi.org/10.1016/j.carbon.2024.119713
Multiscale Simulations on Viscoelastic and Elastoplastic Materials

One of the promising hierarchical modeling for material design is embedding-type multiscale simulation where microscopic simulators (MicS) are embedded in each of the elements of a macroscopic flow/deformation simulation. Figures (a) and (b) show an example of such multiscale simulations for fracture of a solid due to a moving obstacle (gray circle). The deformation of the macroscopic elastic body is described by many coarse-grained particles (smoothed particle hydrodynamics: SPH) shown in Fig.(b). In this model, the local stress-strain relation is evaluated by the MicS’s embedded in each of the SPH particles. When embedding MicS’s into SPH particles, the boundary condition of the MicS plays a crucial role in determining the local stress generated by the macroscopic flow/deformation. Figures (c) and (d) show a successful example of a large elongational deformation under the periodic boundary condition. We introduced UEF (A LAMMPS package for molecular dynamics under extensional flow fields) method and QR decomposition to coarse-grained molecular dynamics (CGMD) simulation and succeeded in simulating a large elongational deformation of polymer melts ( (c) before and (d) after the deformation).
[1] Yohei Morii and Toshihiro Kawakatsu
Phys. Fluids, 33, 9 (2021) Art.No.093106, DOI: https://doi.org/10.1063/5.0063059
[2] Takahiro Murashima, Katsumi Hagita and Toshihiro Kawakatsu
Macromolecules, 54, 15 (2021) pp.7210-7225, DOI: https://doi.org/10.1021/acs.macromol.1c00267
First principles Design of Doping by Impurity Incorporation in 2D Semiconductor MoS2

MoS2 (molybdenum disul de) has attracted significant attention as a next-generation two-dimensional semiconductor material due to its excellent electrical and optical properties. One of the key characteristics of 2D materials is their large surface area per unit volume, which enables higher doping concentrations compared to conventional 3D semiconductors. However, the increased structural flexibility at the surface—such as substitution, adsorption, and interstitial incorporation—makes it difficult to predict stable configurations and electronic properties.
In this study, we systematically analyzed the stable atomic configurations and electronic properties of MoS2 doped with 27 different impurity elements using first-principles calculations. Large-scale computations were performed on the MASAMUNE-IMR supercomputer to comprehensively evaluate the energetic stability of various impurity configurations. The following key findings were obtained:
(1) Stable atomic structures induced by the incorporation of 27 elements were identified, and the carrier conduction type (n-type or p-type) contributed by each impurity was determined.
(2) Including representative dopants such as Re (n-type) and Nb (p-type) in MoS2, it was universally confirmed that all the examined elements form localized electronic states—namely, polaronic states. Moreover, the localized polaron states predicted by the calculations were recently validated by scanning tunneling spectroscopy.
(3) Our results suggest that the conduction mechanism resulting from impurity incorporation predominantly relies on hopping conduction between localized polaron states, providing significant insights into optimizing the device characteristics and understanding the electronic properties of MoS2.
Soungmin Bae, Ibuki Miyamoto, Shin Kiyohara and Yu Kumagai
ACS Nano, 18, 50 (2024) pp.33988-33997, DOI: https://doi.org/10.1021/acsnano.4c08366
Thermal stability of Pd/Sr3Ti2O7 catalyst unveiled by machine learning enhanced global optimization

Self-regenerative materials are keys to the development of stable heterogeneous catalysts used under high-temperature conditions, such as those in three-way catalytic converters in automobiles. Metal nanoparticles supported on perovskite oxides show great promise in this area. However, the atomistic details of these systems, which are vital for understanding and developing thermally stable catalysts, remain largely unexplored.
Herein, we employ a machine-learning-enhanced density functional theory analysis to elucidate the thermal stability of small PdxOy nanoparticles supported on a Sr3Ti2O7 (001) surface. We demonstrate that the PdxOy particles on this support meet the criteria for self-regenerative catalysts. Under oxidative conditions, the solid-solution reaction between Pd and Sr3Ti2O7 is favorable but is limited to the vicinity of the surface. Additionally, the formation of PdO-like clusters strengthens their binding to the support, preventing the agglomeration (sintering) of the PdxOy nanoparticles. Through detailed thermodynamic and electronic structure analyses, we elucidate the roles of the oxide support, the size of the oxidized metal clusters, and the metal-support interaction in the thermal stability of the catalysts. This work paved the way for the rational design of thermally stable catalysts.
T. N. Pham, B. A. C. Tan, Y. Hamamoto, K. Inagaki, I. Hamada and Y. Morikawa
ACS Catal., 14, 3 (2024) pp.1443-1458, DOI: https://doi.org/10.1021/acscatal.3c05673
Access
Access from Sendai Station
walk (20 min)
Taxi (10 min)
Subway (10-15 min)
From Sendai Airport
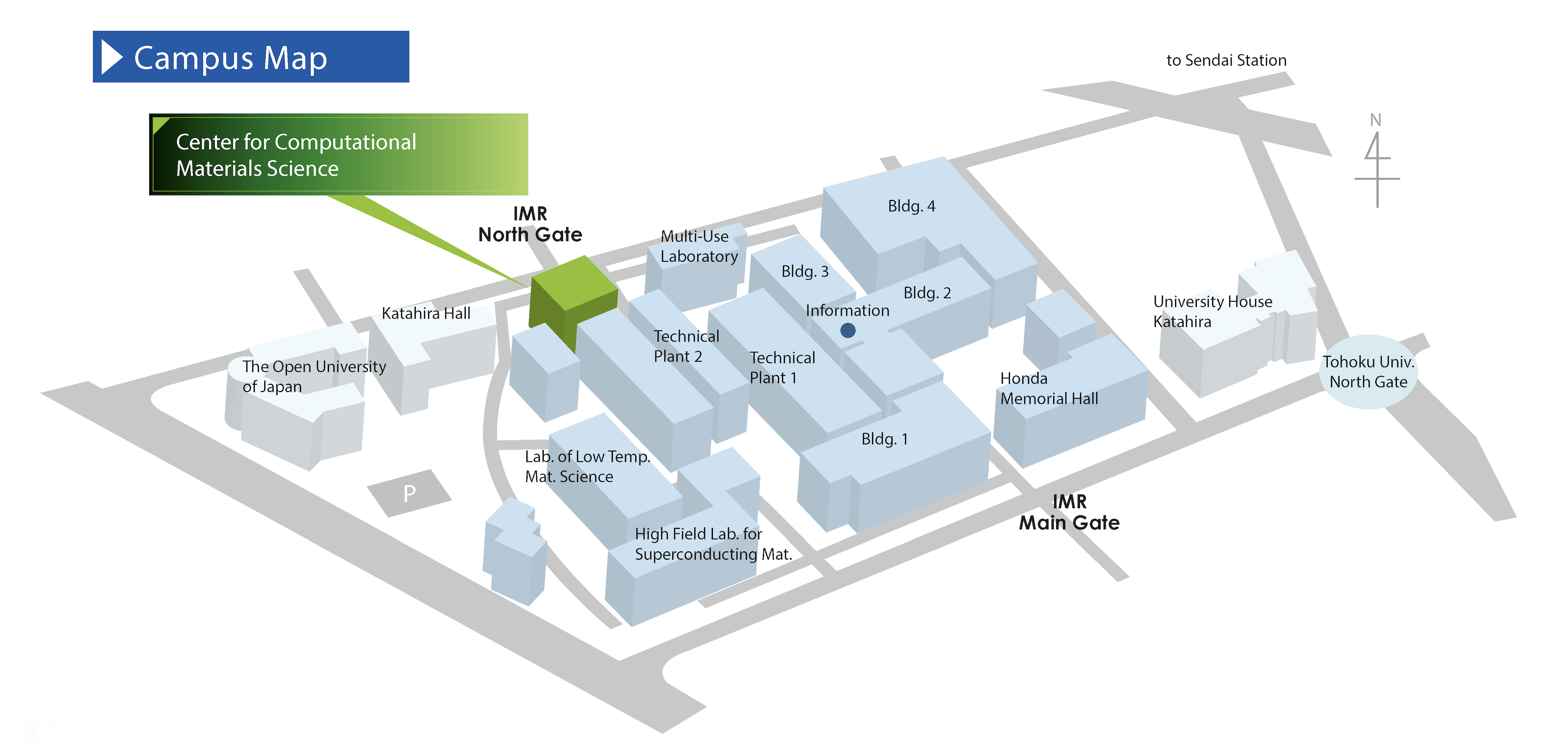
Institute for Materials Research, Tohoku University
Center for Computational Materials Science
TEL: +81-22-215-2411, FAX: +81-22-215-2166

